Abbas E. Kitabchi , Guillermo E. Umpierrez , John M. Miles , Joseph N. Fisher; Hyperglycemic Crises in Adult Patients With Diabetes. Diabetes Care 1 July 2009; 32 (7): 1335–1343. https://doi.org/10.2337/dc09-9032
Download citation file:
toolbar searchDiabetic ketoacidosis (DKA) and the hyperosmolar hyperglycemic state (HHS) are the two most serious acute metabolic complications of diabetes. DKA is responsible for more than 500,000 hospital days per year (1,2) at an estimated annual direct medical expense and indirect cost of 2.4 billion USD (2,3). Table 1 outlines the diagnostic criteria for DKA and HHS. The triad of uncontrolled hyperglycemia, metabolic acidosis, and increased total body ketone concentration characterizes DKA. HHS is characterized by severe hyperglycemia, hyperosmolality, and dehydration in the absence of significant ketoacidosis. These metabolic derangements result from the combination of absolute or relative insulin deficiency and an increase in counterregulatory hormones (glucagon, catecholamines, cortisol, and growth hormone). Most patients with DKA have autoimmune type 1 diabetes; however, patients with type 2 diabetes are also at risk during the catabolic stress of acute illness such as trauma, surgery, or infections. This consensus statement will outline precipitating factors and recommendations for the diagnosis, treatment, and prevention of DKA and HHS in adult subjects. It is based on a previous technical review (4) and more recently published peer-reviewed articles since 2001, which should be consulted for further information.
Diagnostic criteria for DKA and HHS
| . | DKA . | HHS . | ||
|---|---|---|---|---|
| Mild (plasma glucose >250 mg/dl) . | Moderate (plasma glucose >250 mg/dl) . | Severe (plasma glucose >250 mg/dl) . | Plasma glucose >600 mg/dl . | |
| Arterial pH | 7.25–7.30 | 7.00 to | >7.30 | |
| Serum bicarbonate (mEq/l) | 15–18 | 10 to | >18 | |
| Urine ketone* | Positive | Positive | Positive | Small |
| Serum ketone* | Positive | Positive | Positive | Small |
| Effective serum osmolality† | Variable | Variable | Variable | >320 mOsm/kg |
| Anion gap‡ | >10 | >12 | >12 | Variable |
| Mental status | Alert | Alert/drowsy | Stupor/coma | Stupor/coma |
| . | DKA . | HHS . | ||
|---|---|---|---|---|
| Mild (plasma glucose >250 mg/dl) . | Moderate (plasma glucose >250 mg/dl) . | Severe (plasma glucose >250 mg/dl) . | Plasma glucose >600 mg/dl . | |
| Arterial pH | 7.25–7.30 | 7.00 to | >7.30 | |
| Serum bicarbonate (mEq/l) | 15–18 | 10 to | >18 | |
| Urine ketone* | Positive | Positive | Positive | Small |
| Serum ketone* | Positive | Positive | Positive | Small |
| Effective serum osmolality† | Variable | Variable | Variable | >320 mOsm/kg |
| Anion gap‡ | >10 | >12 | >12 | Variable |
| Mental status | Alert | Alert/drowsy | Stupor/coma | Stupor/coma |
*Nitroprusside reaction method.
†Effective serum osmolality: 2[measured Na + (mEq/l)] + glucose (mg/dl)/18.
‡Anion gap: (Na + ) − [(Cl − + HCO3 − (mEq/l)]. (Data adapted from ref. 13.)
Recent epidemiological studies indicate that hospitalizations for DKA in the U.S. are increasing. In the decade from 1996 to 2006, there was a 35% increase in the number of cases, with a total of 136,510 cases with a primary diagnosis of DKA in 2006—a rate of increase perhaps more rapid than the overall increase in the diagnosis of diabetes (1). Most patients with DKA were between the ages of 18 and 44 years (56%) and 45 and 65 years (24%), with only 18% of patients 5% has been reported in the elderly and in patients with concomitant life-threatening illnesses (7,8). Death in these conditions is rarely due to the metabolic complications of hyperglycemia or ketoacidosis but relates to the underlying precipitating illness (4,9). Mortality attributed to HHS is considerably higher than that attributed to DKA, with recent mortality rates of 5–20% (10,11). The prognosis of both conditions is substantially worsened at the extremes of age in the presence of coma, hypotension, and severe comorbidities (1,4,8, 12,13).
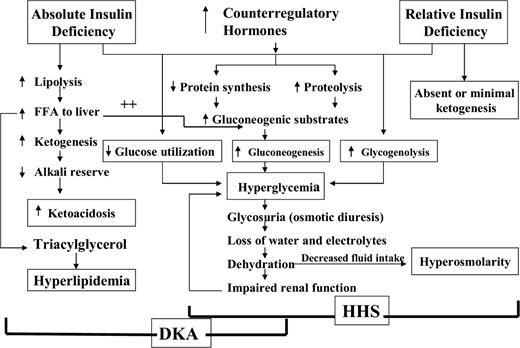
The events leading to hyperglycemia and ketoacidosis are depicted in Fig. 1 (13). In DKA, reduced effective insulin concentrations and increased concentrations of counterregulatory hormones (catecholamines, cortisol, glucagon, and growth hormone) lead to hyperglycemia and ketosis. Hyperglycemia develops as a result of three processes: increased gluconeogenesis, accelerated glycogenolysis, and impaired glucose utilization by peripheral tissues (12, , , , –17). This is magnified by transient insulin resistance due to the hormone imbalance itself as well as the elevated free fatty acid concentrations (4,18). The combination of insulin deficiency and increased counterregulatory hormones in DKA also leads to the release of free fatty acids into the circulation from adipose tissue (lipolysis) and to unrestrained hepatic fatty acid oxidation in the liver to ketone bodies (β-hydroxybutyrate and acetoacetate) (19), with resulting ketonemia and metabolic acidosis.
Pathogenesis of DKA and HHS: stress, infection, or insufficient insulin. FFA, free fatty acid.
Pathogenesis of DKA and HHS: stress, infection, or insufficient insulin. FFA, free fatty acid.
Increasing evidence indicates that the hyperglycemia in patients with hyperglycemic crises is associated with a severe inflammatory state characterized by an elevation of proinflammatory cytokines (tumor necrosis factor-α and interleukin-β, -6, and -8), C-reactive protein, reactive oxygen species, and lipid peroxidation, as well as cardiovascular risk factors, plasminogen activator inhibitor-1 and free fatty acids in the absence of obvious infection or cardiovascular pathology (20). All of these parameters return to near-normal values with insulin therapy and hydration within 24 h. The procoagulant and inflammatory states may be due to nonspecific phenomena of stress and may partially explain the association of hyperglycemic crises with a hypercoagulable state (21).
The pathogenesis of HHS is not as well understood as that of DKA, but a greater degree of dehydration (due to osmotic diuresis) and differences in insulin availability distinguish it from DKA (4,22). Although relative insulin deficiency is clearly present in HHS, endogenous insulin secretion (reflected by C-peptide levels) appears to be greater than in DKA, where it is negligible (Table 2). Insulin levels in HHS are inadequate to facilitate glucose utilization by insulin-sensitive tissues but adequate to prevent lipolysis and subsequent ketogenesis (12).
Admission biochemical data in patients with HHS or DKA
| . | HHS . | DKA . |
|---|---|---|
| Glucose (mg/dl) | 930 ± 83 | 616 ± 36 |
| Na + (mEq/l) | 149 ± 3.2 | 134 ± 1.0 |
| K + (mEq/l) | 3.9 ± 0.2 | 4.5 ± 0.13 |
| BUN (mg/dl) | 61 ± 11 | 32 ± 3 |
| Creatinine (mg/dl) | 1.4 ± 0.1 | 1.1 ± 0.1 |
| pH | 7.3 ± 0.03 | 7.12 ± 0.04 |
| Bicarbonate (mEq/l) | 18 ± 1.1 | 9.4 ± 1.4 |
| 3-β-hydroxybutyrate (mmol/l) | 1.0 ± 0.2 | 9.1 ± 0.85 |
| Total osmolality* | 380 ± 5.7 | 323 ± 2.5 |
| IRI (nmol/l) | 0.08 ± 0.01 | 0.07 ± 0.01 |
| C-peptide (nmol/l) | 1.14 ± 0.1 | 0.21 ± 0.03 |
| Free fatty acids (nmol/l) | 1.5 ± 0.19 | 1.6 ± 0.16 |
| Human growth hormone (ng/ml) | 1.9 ± 0.2 | 6.1 ± 1.2 |
| Cortisol (ng/ml) | 570 ± 49 | 500 ± 61 |
| IRI (nmol/l)† | 0.27 ± 0.05 | 0.09 ± 0.01 |
| C-peptide (nmol/l)† | 1.75 ± 0.23 | 0.25 ± 0.05 |
| Glucagon (ng/ml) | 689 ± 215 | 580 ± 147 |
| Catacholamines (ng/ml) | 0.28 ± 0.09 | 1.78 ± 0.4 |
| Growth hormone (ng/ml) | 1.1 | 7.9 |
| ΔGap: anion gap − 12 (mEq/l) | 11 | 17 |
| . | HHS . | DKA . |
|---|---|---|
| Glucose (mg/dl) | 930 ± 83 | 616 ± 36 |
| Na + (mEq/l) | 149 ± 3.2 | 134 ± 1.0 |
| K + (mEq/l) | 3.9 ± 0.2 | 4.5 ± 0.13 |
| BUN (mg/dl) | 61 ± 11 | 32 ± 3 |
| Creatinine (mg/dl) | 1.4 ± 0.1 | 1.1 ± 0.1 |
| pH | 7.3 ± 0.03 | 7.12 ± 0.04 |
| Bicarbonate (mEq/l) | 18 ± 1.1 | 9.4 ± 1.4 |
| 3-β-hydroxybutyrate (mmol/l) | 1.0 ± 0.2 | 9.1 ± 0.85 |
| Total osmolality* | 380 ± 5.7 | 323 ± 2.5 |
| IRI (nmol/l) | 0.08 ± 0.01 | 0.07 ± 0.01 |
| C-peptide (nmol/l) | 1.14 ± 0.1 | 0.21 ± 0.03 |
| Free fatty acids (nmol/l) | 1.5 ± 0.19 | 1.6 ± 0.16 |
| Human growth hormone (ng/ml) | 1.9 ± 0.2 | 6.1 ± 1.2 |
| Cortisol (ng/ml) | 570 ± 49 | 500 ± 61 |
| IRI (nmol/l)† | 0.27 ± 0.05 | 0.09 ± 0.01 |
| C-peptide (nmol/l)† | 1.75 ± 0.23 | 0.25 ± 0.05 |
| Glucagon (ng/ml) | 689 ± 215 | 580 ± 147 |
| Catacholamines (ng/ml) | 0.28 ± 0.09 | 1.78 ± 0.4 |
| Growth hormone (ng/ml) | 1.1 | 7.9 |
| ΔGap: anion gap − 12 (mEq/l) | 11 | 17 |
*According to the formula 2(Na + K) + urea (mmol/l) + glucose (mmol/l).
†Values following intravenous administration of tolbutamide. IRI, immunoreactive insulin. (Adapted from ref. 4.)
The most common precipitating factor in the development of DKA and HHS is infection (1,4,10). Other precipitating factors include discontinuation of or inadequate insulin therapy, pancreatitis, myocardial infarction, cerebrovascular accident, and drugs (10,13,14). In addition, new-onset type 1 diabetes or discontinuation of insulin in established type 1 diabetes commonly leads to the development of DKA. In young patients with type 1 diabetes, psychological problems complicated by eating disorders may be a contributing factor in 20% of recurrent ketoacidosis. Factors that may lead to insulin omission in younger patients include fear of weight gain with improved metabolic control, fear of hypoglycemia, rebellion against authority, and stress of chronic disease.
Before 1993, the use of continuous subcutaneous insulin infusion devices had also been associated with an increased frequency of DKA (23); however, with improvement in technology and better education of patients, the incidence of DKA appears to have reduced in pump users. However, additional prospective studies are needed to document reduction of DKA incidence with the use of continuous subcutaneous insulin infusion devices (24).
Underlying medical illness that provokes the release of counterregulatory hormones or compromises the access to water is likely to result in severe dehydration and HHS. In most patients with HHS, restricted water intake is due to the patient being bedridden and is exacerbated by the altered thirst response of the elderly. Because 20% of these patients have no history of diabetes, delayed recognition of hyperglycemic symptoms may have led to severe dehydration. Elderly individuals with new-onset diabetes (particularly residents of chronic care facilities) or individuals with known diabetes who become hyperglycemic and are unaware of it or are unable to take fluids when necessary are at risk for HHS (10,25).
Drugs that affect carbohydrate metabolism, such as corticosteroids, thiazides, sympathomimetic agents, and pentamidine, may precipitate the development of HHS or DKA (4). Recently, a number of case reports indicate that the conventional antipsychotic as well as atypical antipsychotic drugs may cause hyperglycemia and even DKA or HHS (26,27). Possible mechanisms include the induction of peripheral insulin resistance and the direct influence on pancreatic β-cell function by 5-HT1A/2A/2C receptor antagonism, by inhibitory effects via α2-adrenergic receptors, or by toxic effects (28).
An increasing number of DKA cases without precipitating cause have been reported in children, adolescents, and adult subjects with type 2 diabetes. Observational and prospective studies indicate that over half of newly diagnosed adult African American and Hispanic subjects with unprovoked DKA have type 2 diabetes (28, , , –32). The clinical presentation in such cases is acute (as in classical type 1 diabetes); however, after a short period of insulin therapy, prolonged remission is often possible, with eventual cessation of insulin treatment and maintenance of glycemic control with diet or oral antihyperglycemic agents. In such patients, clinical and metabolic features of type 2 diabetes include a high rate of obesity, a strong family history of diabetes, a measurable pancreatic insulin reserve, a low prevalence of autoimmune markers of β-cell destruction, and the ability to discontinue insulin therapy during follow-up (28, 31,32). This unique, transient insulin-requiring profile after DKA has been recognized mainly in blacks and Hispanics but has also been reported in Native American, Asian, and white populations (32). This variant of diabetes has been referred to in the literature as idiopathic type 1 diabetes, atypical diabetes, “Flatbush diabetes,” type 1.5 diabetes, and more recently, ketosis-prone type 2 diabetes. Some experimental work has shed a mechanistic light on the pathogenesis of ketosis-prone type 2 diabetes. At presentation, they have markedly impaired insulin secretion and insulin action, but aggressive management with insulin improves insulin secretion and action to levels similar to those of patients with type 2 diabetes without DKA (28,31,32). Recently, it has been reported that the near-normoglycemic remission is associated with a greater recovery of basal and stimulated insulin secretion and that 10 years after diabetes onset, 40% of patients are still non–insulin dependent (31). Fasting C-peptide levels of >1.0 ng/dl (0.33 nmol/l) and stimulated C-peptide levels >1.5 ng/dl (0.5 nmol/l) are predictive of long-term normoglycemic remission in patients with a history of DKA (28,32).
The process of HHS usually evolves over several days to weeks, whereas the evolution of the acute DKA episode in type 1 diabetes or even in type 2 diabetes tends to be much shorter. Although the symptoms of poorly controlled diabetes may be present for several days, the metabolic alterations typical of ketoacidosis usually evolve within a short time frame (typically 50%) but are uncommon in HHS (33). Caution needs to be taken with patients who complain of abdominal pain on presentation because the symptoms could be either a result of the DKA or an indication of a precipitating cause of DKA, particularly in younger patients or in the absence of severe metabolic acidosis (34,35). Further evaluation is necessary if this complaint does not resolve with resolution of dehydration and metabolic acidosis.
The diagnostic criteria for DKA and HHS are shown in Table 1. The initial laboratory evaluation of patients include determination of plasma glucose, blood urea nitrogen, creatinine, electrolytes (with calculated anion gap), osmolality, serum and urinary ketones, and urinalysis, as well as initial arterial blood gases and a complete blood count with a differential. An electrocardiogram, chest X-ray, and urine, sputum, or blood cultures should also be obtained.
The severity of DKA is classified as mild, moderate, or severe based on the severity of metabolic acidosis (blood pH, bicarbonate, and ketones) and the presence of altered mental status (4). Significant overlap between DKA and HHS has been reported in more than one-third of patients (36). Although most patients with HHS have an admission pH >7.30 and a bicarbonate level >18 mEq/l, mild ketonemia may be present (4,10).
Severe hyperglycemia and dehydration with altered mental status in the absence of significant acidosis characterize HHS, which clinically presents with less ketosis and greater hyperglycemia than DKA. This may result from a plasma insulin concentration (as determined by baseline and stimulated C-peptide [Table 2]) adequate to prevent excessive lipolysis and subsequent ketogenesis but not hyperglycemia (4).
The key diagnostic feature in DKA is the elevation in circulating total blood ketone concentration. Assessment of augmented ketonemia is usually performed by the nitroprusside reaction, which provides a semiquantitative estimation of acetoacetate and acetone levels. Although the nitroprusside test (both in urine and in serum) is highly sensitive, it can underestimate the severity of ketoacidosis because this assay does not recognize the presence of β-hydroxybutyrate, the main metabolic product in ketoacidosis (4,12). If available, measurement of serum β-hydroxybutyrate may be useful for diagnosis (37). Accumulation of ketoacids results in an increased anion gap metabolic acidosis. The anion gap is calculated by subtracting the sum of chloride and bicarbonate concentration from the sodium concentration: [Na − (Cl + HCO3)]. A normal anion gap is between 7 and 9 mEq/l and an anion gap >10–12 mEq/l indicate the presence of increased anion gap metabolic acidosis (4).
Hyperglycemia is a key diagnostic criterion of DKA; however, a wide range of plasma glucose can be present on admission. Elegant studies on hepatic glucose production rates have reported rates ranging from normal or near normal (38) to elevated (12,15), possibly contributing to the wide range of plasma glucose levels in DKA that are independent of the severity of ketoacidosis (37). Approximately 10% of the DKA population presents with so-called “euglycemic DKA”—glucose levels ≤250 mg/dl (38). This could be due to a combination of factors, including exogenous insulin injection en route to the hospital, antecedent food restriction (39, 40), and inhibition of gluconeogenesis.
On admission, leukocytosis with cell counts in the 10,000–15,000 mm 3 range is the rule in DKA and may not be indicative of an infectious process. However, leukocytosis with cell counts >25,000 mm 3 may designate infection and require further evaluation (41). In ketoacidosis, leukocytosis is attributed to stress and maybe correlated to elevated levels of cortisol and norepinephrine (42). The admission serum sodium is usually low because of the osmotic flux of water from the intracellular to the extracellular space in the presence of hyperglycemia. An increased or even normal serum sodium concentration in the presence of hyperglycemia indicates a rather profound degree of free water loss. To assess the severity of sodium and water deficit, serum sodium may be corrected by adding 1.6 mg/dl to the measured serum sodium for each 100 mg/dl of glucose above 100 mg/dl (4,12).
Studies on serum osmolality and mental alteration have established a positive linear relationship between osmolality and mental obtundation (9,36). The occurrence of stupor or coma in a diabetic patient in the absence of definitive elevation of effective osmolality (≥320 mOsm/kg) demands immediate consideration of other causes of mental status change. In the calculation of effective osmolality, [sodium ion (mEq/l) × 2 + glucose (mg/dl)/18], the urea concentration is not taken into account because it is freely permeable and its accumulation does not induce major changes in intracellular volume or osmotic gradient across the cell membrane (4).
Serum potassium concentration may be elevated because of an extracellular shift of potassium caused by insulin deficiency, hypertonicity, and acidemia (43). Patients with low normal or low serum potassium concentration on admission have severe total-body potassium deficiency and require careful cardiac monitoring and more vigorous potassium replacement because treatment lowers potassium further and can provoke cardiac dysrhythmia. Pseudonormoglycemia (44) and pseudohyponatremia (45) may occur in DKA in the presence of severe chylomicronemia.
The admission serum phosphate level in patients with DKA, like serum potassium, is usually elevated and does not reflect an actual body deficit that uniformly exists due to shifts of intracellular phosphate to the extracellular space (12, 46,47). Insulin deficiency, hypertonicity, and increased catabolism all contribute to the movement of phosphate out of cells.
Hyperamylasemia has been reported in 21–79% of patients with DKA (48); however, there is little correlation between the presence, degree, or isoenzyme type of hyperamylasemia and the presence of gastrointestinal symptoms (nausea, vomiting, and abdominal pain) or pancreatic imaging studies (48). A serum lipase determination may be beneficial in the differential diagnosis of pancreatitis; however, lipase could also be elevated in DKA in the absence of pancreatitis (48).
Not all patients with ketoacidosis have DKA. Starvation ketosis and alcoholic ketoacidosis are distinguished by clinical history and by plasma glucose concentrations that range from mildly elevated (rarely >200 mg/dl) to hypoglycemia (49). In addition, although alcoholic ketoacidosis can result in profound acidosis, the serum bicarbonate concentration in starvation ketosis is usually not
A clinical history of previous drug abuse should be sought. Measurement of serum salicylate and blood methanol level may be helpful. Ethylene glycol (antifreeze) is suggested by the presence of calcium oxalate and hippurate crystals in the urine. Paraldehyde ingestion is indicated by its characteristic strong odor on the breath. Because these intoxicants are low–molecular weight organic compounds, they can produce an osmolar gap in addition to the anion gap acidosis (14). A recent report states that active cocaine use is an independent risk factor for recurrent DKA (50).
Recently, one case report has shown that a patient with diagnosed acromegaly may present with DKA as the primary manifestation of the disease (51). In addition, an earlier report of pituitary gigantism was presented with two episodes of DKA with complete resolution of diabetes after pituitary apoplexy (52).
Successful treatment of DKA and HHS requires correction of dehydration, hyperglycemia, and electrolyte imbalances; identification of comorbid precipitating events; and above all, frequent patient monitoring. Protocols for the management of patients with DKA and HHS are summarized in Fig. 2 (52).
Protocol for management of adult patients with DKA or HHS. DKA diagnostic criteria: blood glucose 250 mg/dl, arterial pH 7.3, bicarbonate 15 mEq/l, and moderate ketonuria or ketonemia. HHS diagnostic criteria: serum glucose >600 mg/dl, arterial pH >7.3, serum bicarbonate >15 mEq/l, and minimal ketonuria and ketonemia. †15–20 ml/kg/h; ‡serum Na should be corrected for hyperglycemia (for each 100 mg/dl glucose 100 mg/dl, add 1.6 mEq to sodium value for corrected serum value). (Adapted from ref. 13.) Bwt, body weight; IV, intravenous; SC, subcutaneous.
Protocol for management of adult patients with DKA or HHS. DKA diagnostic criteria: blood glucose 250 mg/dl, arterial pH 7.3, bicarbonate 15 mEq/l, and moderate ketonuria or ketonemia. HHS diagnostic criteria: serum glucose >600 mg/dl, arterial pH >7.3, serum bicarbonate >15 mEq/l, and minimal ketonuria and ketonemia. †15–20 ml/kg/h; ‡serum Na should be corrected for hyperglycemia (for each 100 mg/dl glucose 100 mg/dl, add 1.6 mEq to sodium value for corrected serum value). (Adapted from ref. 13.) Bwt, body weight; IV, intravenous; SC, subcutaneous.
Initial fluid therapy is directed toward expansion of the intravascular, interstitial, and intracellular volume, all of which are reduced in hyperglycemic crises (53) and restoration of renal perfusion. In the absence of cardiac compromise, isotonic saline (0.9% NaCl) is infused at a rate of 15–20 ml · kg body wt −1 · h −1 or 1–1.5 l during the first hour. Subsequent choice for fluid replacement depends on hemodynamics, the state of hydration, serum electrolyte levels, and urinary output. In general, 0.45% NaCl infused at 250–500 ml/h is appropriate if the corrected serum sodium is normal or elevated; 0.9% NaCl at a similar rate is appropriate if corrected serum sodium is low (Fig. 2). Successful progress with fluid replacement is judged by hemodynamic monitoring (improvement in blood pressure), measurement of fluid input/output, laboratory values, and clinical examination. Fluid replacement should correct estimated deficits within the first 24 h. In patients with renal or cardiac compromise, monitoring of serum osmolality and frequent assessment of cardiac, renal, and mental status must be performed during fluid resuscitation to avoid iatrogenic fluid overload (4,10, 15,53). Aggressive rehydration with subsequent correction of the hyperosmolar state has been shown to result in a more robust response to low-dose insulin therapy (54).
During treatment of DKA, hyperglycemia is corrected faster than ketoacidosis. The mean duration of treatment until blood glucose is 7.30; bicarbonate >18 mmol/l) is corrected is 6 and 12 h, respectively (9,55). Once the plasma glucose is ∼ 200 mg/dl, 5% dextrose should be added to replacement fluids to allow continued insulin administration until ketonemia is controlled while at the same time avoiding hypoglycemia.
The mainstay in the treatment of DKA involves the administration of regular insulin via continuous intravenous infusion or by frequent subcutaneous or intramuscular injections (4,56,57). Randomized controlled studies in patients with DKA have shown that insulin therapy is effective regardless of the route of administration (47). The administration of continuous intravenous infusion of regular insulin is the preferred route because of its short half-life and easy titration and the delayed onset of action and prolonged half-life of subcutaneous regular insulin (36,47,58).
Numerous prospective randomized studies have demonstrated that use of low-dose regular insulin by intravenous infusion is sufficient for successful recovery of patients with DKA. Until recently, treatment algorithms recommended the administration of an initial intravenous dose of regular insulin (0.1 units/kg) followed by the infusion of 0.1 units · kg −1 · h −1 insulin (Fig. 2). A recent prospective randomized study reported that a bolus dose of insulin is not necessary if patients receive an hourly insulin infusion of 0.14 units/kg body wt (equivalent to 10 units/h in a 70-kg patient) (59). In the absence of an initial bolus, however, doses
Low-dose insulin infusion protocols decrease plasma glucose concentration at a rate of 50–75 mg · dl −1 · h −1 . If plasma glucose does not decrease by 50–75 mg from the initial value in the first hour, the insulin infusion should be increased every hour until a steady glucose decline is achieved (Fig. 2). When the plasma glucose reaches 200 mg/dl in DKA or 300 mg/dl in HHS, it may be possible to decrease the insulin infusion rate to 0.02– 0.05 units · kg −1 · h −1 , at which time dextrose may be added to the intravenous fluids (Fig. 2). Thereafter, the rate of insulin administration or the concentration of dextrose may need to be adjusted to maintain glucose values between 150 and 200 mg/dl in DKA or 250 and 300 mg/dl in HHS until they are resolved.
Treatment with subcutaneous rapid-acting insulin analogs (lispro and aspart) has been shown to be an effective alternative to the use of intravenous regular insulin in the treatment of DKA. Treatment of patients with mild and moderate DKA with subcutaneous rapid-acting insulin analogs every 1 or 2 h in non–intensive care unit (ICU) settings has been shown to be as safe and effective as the treatment with intravenous regular insulin in the ICU (60,61). The rate of decline of blood glucose concentration and the mean duration of treatment until correction of ketoacidosis were similar among patients treated with subcutaneous insulin analogs every 1 or 2 h or with intravenous regular insulin. However, until these studies are confirmed outside the research arena, patients with severe DKA, hypotension, anasarca, or associated severe critical illness should be managed with intravenous regular insulin in the ICU.
Despite total-body potassium depletion, mild-to-moderate hyperkalemia is common in patients with hyperglycemic crises. Insulin therapy, correction of acidosis, and volume expansion decrease serum potassium concentration. To prevent hypokalemia, potassium replacement is initiated after serum levels fall below the upper level of normal for the particular laboratory (5.0–5.2 mEq/l). The treatment goal is to maintain serum potassium levels within the normal range of 4–5 mEq/l. Generally, 20–30 mEq potassium in each liter of infusion fluid is sufficient to maintain a serum potassium concentration within the normal range. Rarely, DKA patients may present with significant hypokalemia. In such cases, potassium replacement should begin with fluid therapy, and insulin treatment should be delayed until potassium concentration is restored to >3.3 mEq/l to avoid life-threatening arrhythmias and respiratory muscle weakness (4,13).
The use of bicarbonate in DKA is controversial (62) because most experts believe that during the treatment, as ketone bodies decrease there will be adequate bicarbonate except in severely acidotic patients. Severe metabolic acidosis can lead to impaired myocardial contractility, cerebral vasodilatation and coma, and several gastrointestinal complications (63). A prospective randomized study in 21 patients failed to show either beneficial or deleterious changes in morbidity or mortality with bicarbonate therapy in DKA patients with an admission arterial pH between 6.9 and 7.1 (64). Nine small studies in a total of 434 patients with diabetic ketoacidosis (217 treated with bicarbonate and 178 patients without alkali therapy [(62)]) support the notion that bicarbonate therapy for DKA offers no advantage in improving cardiac or neurologic functions or in the rate of recovery of hyperglycemia and ketoacidosis. Moreover, several deleterious effects of bicarbonate therapy have been reported, such as increased risk of hypokalemia, decreased tissue oxygen uptake (65), cerebral edema (65), and development of paradoxical central nervous system acidosis.
Despite whole-body phosphate deficits in DKA that average 1.0 mmol/kg body wt, serum phosphate is often normal or increased at presentation. Phosphate concentration decreases with insulin therapy. Prospective randomized studies have failed to show any beneficial effect of phosphate replacement on the clinical outcome in DKA (46,67), and overzealous phosphate therapy can cause severe hypocalcemia (46,68). However, to avoid potential cardiac and skeletal muscle weakness and respiratory depression due to hypophosphatemia, careful phosphate replacement may sometimes be indicated in patients with cardiac dysfunction, anemia, or respiratory depression and in those with serum phosphate concentration 2 PO4) (69). No studies are available on the use of phosphate in the treatment of HHS.
Patients with DKA and HHS should be treated with continuous intravenous insulin until the hyperglycemic crisis is resolved. Criteria for resolution of ketoacidosis include a blood glucose 7.3, and a calculated anion gap ≤12 mEq/l. Resolution of HHS is associated with normal osmolality and regain of normal mental status. When this occurs, subcutaneous insulin therapy can be started. To prevent recurrence of hyperglycemia or ketoacidosis during the transition period to subcutaneous insulin, it is important to allow an overlap of 1–2 h between discontinuation of intravenous insulin and the administration of subcutaneous insulin. If the patient is to remain fasting/nothing by mouth, it is preferable to continue the intravenous insulin infusion and fluid replacement. Patients with known diabetes may be given insulin at the dosage they were receiving before the onset of DKA so long as it was controlling glucose properly. In insulin-naïve patients, a multidose insulin regimen should be started at a dose of 0.5–0.8 units · kg −1 · day −1 (13). Human insulin (NPH and regular) are usually given in two or three doses per day. More recently, basal-bolus regimens with basal (glargine and detemir) and rapid-acting insulin analogs (lispro, aspart, or glulisine) have been proposed as a more physiologic insulin regimen in patients with type 1 diabetes. A prospective randomized trial compared treatment with a basal-bolus regimen, including glargine once daily and glulisine before meals, with a split-mixed regimen of NPH plus regular insulin twice daily following the resolution of DKA. Transition to subcutaneous glargine and glulisine resulted in similar glycemic control compared with NPH and regular insulin; however, treatment with basal bolus was associated with a lower rate of hypoglycemic events (15%) than the rate in those treated with NPH and regular insulin (41%) (55).
Hypoglycemia and hypokalemia are two common complications with overzealous treatment of DKA with insulin and bicarbonate, respectively, but these complications have occurred less often with the low-dose insulin therapy (4,56,57). Frequent blood glucose monitoring (every 1–2 h) is mandatory to recognize hypoglycemia because many patients with DKA who develop hypoglycemia during treatment do not experience adrenergic manifestations of sweating, nervousness, fatigue, hunger, and tachycardia. Hyperchloremic non–anion gap acidosis, which is seen during the recovery phase of DKA, is self-limited with few clinical consequences (43). This may be caused by loss of ketoanions, which are metabolized to bicarbonate during the evolution of DKA and excess fluid infusion of chloride containing fluids during treatment (4).
Cerebral edema, which occurs in ∼0.3–1.0% of DKA episodes in children, is extremely rare in adult patients during treatment of DKA. Cerebral edema is associated with a mortality rate of 20–40% (5) and accounts for 57–87% of all DKA deaths in children (70,71). Symptoms and signs of cerebral edema are variable and include onset of headache, gradual deterioration in level of consciousness, seizures, sphincter incontinence, pupillary changes, papilledema, bradycardia, elevation in blood pressure, and respiratory arrest (71). A number of mechanisms have been proposed, which include the role of cerebral ischemia/hypoxia, the generation of various inflammatory mediators (72), increased cerebral blood flow, disruption of cell membrane ion transport, and a rapid shift in extracellular and intracellular fluids resulting in changes in osmolality. Prevention might include avoidance of excessive hydration and rapid reduction of plasma osmolarity, a gradual decrease in serum glucose, and maintenance of serum glucose between 250–300 mg/dl until the patient's serum osmolality is normalized and mental status is improved. Manitol infusion and mechanical ventilation are suggested for treatment of cerebral edema (73).
Many cases of DKA and HHS can be prevented by better access to medical care, proper patient education, and effective communication with a health care provider during an intercurrent illness. Paramount in this effort is improved education regarding sick day management, which includes the following:
The use of home glucose-ketone meters may allow early recognition of impending ketoacidosis, which may help to guide insulin therapy at home and, possibly, may prevent hospitalization for DKA. In addition, home blood ketone monitoring, which measures β-hydroxybutyrate levels on a fingerstick blood specimen, is now commercially available (37).
The observation that stopping insulin for economic reasons is a common precipitant of DKA (74,75) underscores the need for our health care delivery systems to address this problem, which is costly and clinically serious. The rate of insulin discontinuation and a history of poor compliance accounts for more than half of DKA admissions in inner-city and minority populations (9,74,75). Several cultural and socioeconomic barriers, such as low literacy rate, limited financial resources, and limited access to health care, in medically indigent patients may explain the lack of compliance and why DKA continues to occur in such high rates in inner-city patients. These findings suggest that the current mode of providing patient education and health care has significant limitations. Addressing health problems in the African American and other minority communities requires explicit recognition of the fact that these populations are probably quite diverse in their behavioral responses to diabetes (76).
Significant resources are spent on the cost of hospitalization. DKA episodes represent >1 of every 4 USD spent on direct medical care for adult patients with type 1 diabetes and 1 of every 2 USD in patients experiencing multiple episodes (77). Based on an annual average of 135,000 hospitalizations for DKA in the U.S., with an average cost of 17,500 USD per patient, the annual hospital cost for patients with DKA may exceed 2.4 billion USD per year (3). A recent study (2) reported that the cost burden resulting from avoidable hospitalizations due to short-term uncontrolled diabetes including DKA is substantial (2.8 billion USD). However, the long-term impact of uncontrolled diabetes and its economic burden could be more significant because it can contribute to various complications. Because most cases occur in patients with known diabetes and with previous DKA, resources need to be redirected toward prevention by funding better access to care and educational programs tailored to individual needs, including ethnic and personal health care beliefs. In addition, resources should be directed toward the education of primary care providers and school personnel so that they can identify signs and symptoms of uncontrolled diabetes and so that new-onset diabetes can be diagnosed at an earlier time. Recent studies suggest that any type of education for nutrition has resulted in reduced hospitalization (78). In fact, the guidelines for diabetes self-management education were developed by a recent task force to identify ten detailed standards for diabetes self-management education (79).
An American Diabetes Association consensus statement represents the authors' collective analysis, evaluation, and opinion at the time of publication and does not represent official association opinion.